Publications
The publications listed here are those affiliated with CompBioClub. For complete lists, see the members' scholar pages.
-
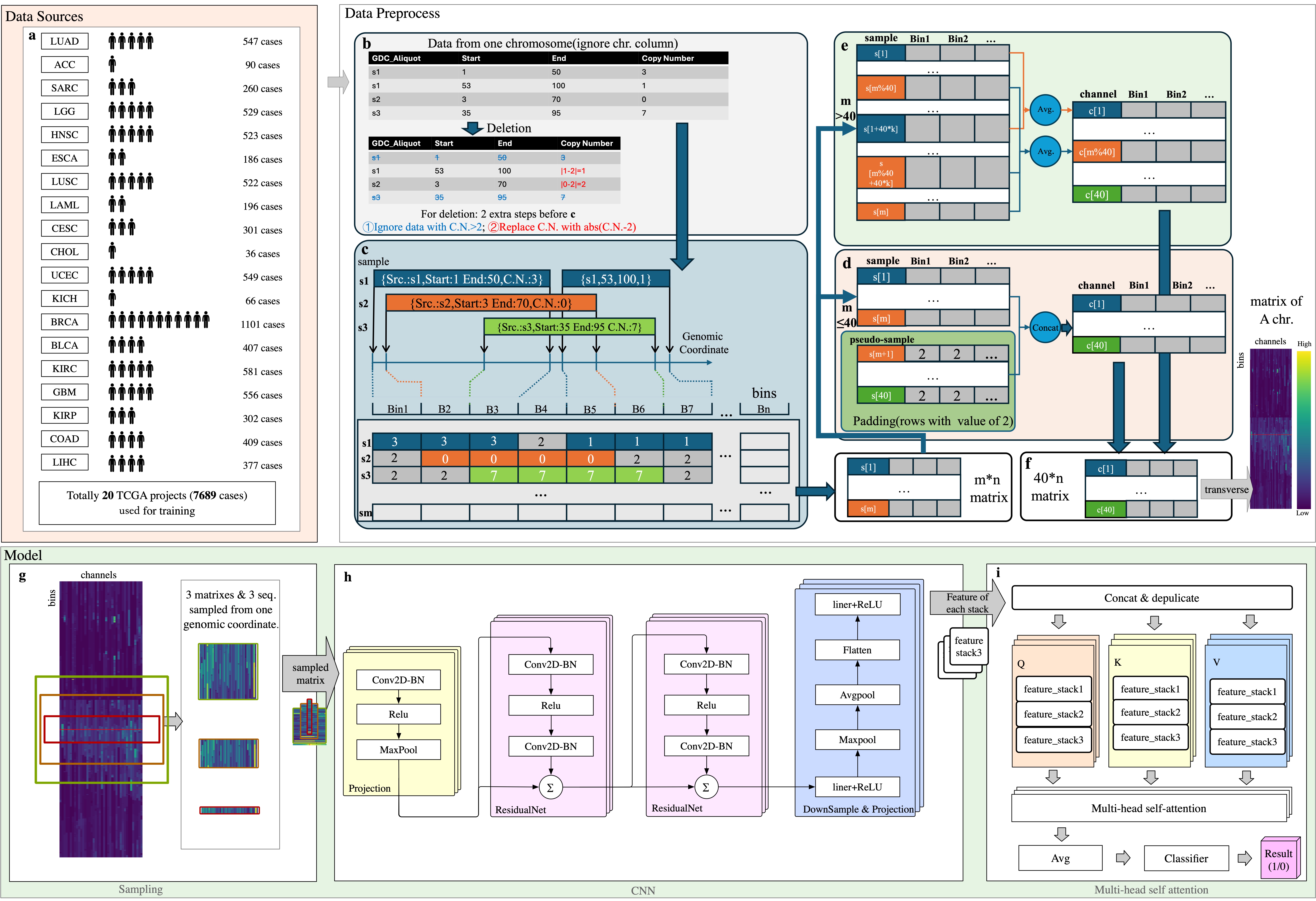 CRESCENT: A Deep Learning Framework with Multi-Scale Attention for Detecting Recurrent Copy Number AberrationsXikang Feng†*, Zheng Xu†, Sisi Peng, Jieyi Zheng, Chuan Ma, Qiangguo Jin*, and Lingxi Chen*Briefings In Bioinformatics, 2026
CRESCENT: A Deep Learning Framework with Multi-Scale Attention for Detecting Recurrent Copy Number AberrationsXikang Feng†*, Zheng Xu†, Sisi Peng, Jieyi Zheng, Chuan Ma, Qiangguo Jin*, and Lingxi Chen*Briefings In Bioinformatics, 2026Recurrent copy number alterations (CNAs) are fundamental drivers of tumorigenesis, yet identifying them reliably remains a challenge due to the extreme variability in their genomic scale and context. Current methods often struggle to balance sensitivity across focal, segmental, and arm-level events. Here, we present CRESCENT, a deep learning framework designed to detect recurrent CNAs by integrating multi-scale sampling with convolutional neural networks and self-attention mechanisms. By processing copy number profiles from 7,689 cases across 20 TCGA cancer projects, CRESCENT learns to distinguish recurrent drivers from background noise through parallel feature fusion. In rigorous leave-one-project-out cross-validation, the model demonstrated robust generalization, achieving AUCs of 0.894-0.967 for amplifications and 0.804-0.929 for deletions in representative cohorts (BLCA, SARC, GBM, UCEC). Finally, extending beyond the TCGA-specific cross-validation, we trained a unified pan-cancer model to assess CRESCENT’s generalizability on simulated datasets and independent, non-TCGA cancer cohorts (CGCI and TARGET). Benchmarking against standard tools, including GISTIC2 and RUBIC, reveals that CRESCENT offers superior detection balance, identifying the highest total number of significant events across focal and broad scales. Moreover, extensive focal gene expression validation and pathway annotation, coupled with survival analysis, highlights that CRESCENT identifies critical oncogenic drivers and prognostic markers that conventional statistical methods often overlook. In all, CRESCENT provides a highly sensitive, generalized approach for decoding tumor evolution.
@article{CRESCENT, title = {CRESCENT: A Deep Learning Framework with Multi-Scale Attention for Detecting Recurrent Copy Number Aberrations}, author = {Feng, Xikang and Xu, Zheng and Peng, Sisi and Zheng, Jieyi and Ma, Chuan and Jin, Qiangguo and Chen, Lingxi}, journal = {Briefings In Bioinformatics}, pages = {2026--03}, year = {2026}, publisher = {Oxford University Press}, doi = {10.1093/bib/bbag167}, peerreviewed = {true} } -
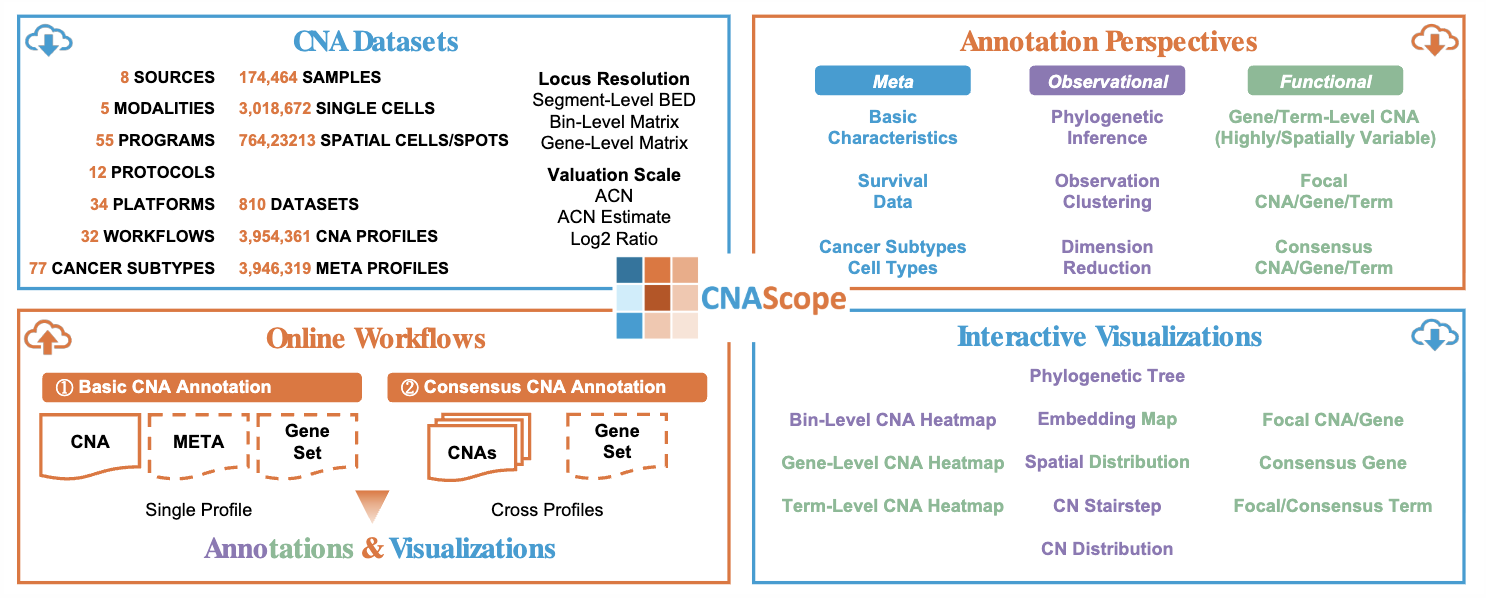 CNAScope: Pan-Cancer Copy Number Aberration Database with Functional Annotation and Interactive VisualizationXikang Feng†*, Jieyi Zheng†, Sisi Peng†, Anna Jiang†, Ka Ho Ng, Chengshang Lyu, Qiangguo Jin*, and Lingxi Chen*Nucleic Acids Research, 2026
CNAScope: Pan-Cancer Copy Number Aberration Database with Functional Annotation and Interactive VisualizationXikang Feng†*, Jieyi Zheng†, Sisi Peng†, Anna Jiang†, Ka Ho Ng, Chengshang Lyu, Qiangguo Jin*, and Lingxi Chen*Nucleic Acids Research, 2026Copy number aberrations (CNAs) are critical drivers of genomic diversity in oncology, where recurrent CNAs frequently underlie tumorigenesis. However, existing public resources are limited in their somatic CNA specificity, breadth across multiple data modalities, and support for recurrent CNAs with online functional annotation and interactive visualization. Here, we present CNAScope (https://cna.compbio.com/), a database that curates and functionally annotates over 3,954,361 CNA profiles and 3,946,319 metadata from 810 datasets, 174,464 samples, 3,018,672 single cells, and 764,232 spatial cells/spots, spanning 77 cancer subtypes from eight data sources and 55 cancer initiatives and institutions. CNAScope offers downloadable CNA annotations and interactive visualizations at bin, gene, and pathway term levels, including phylogenetic inference, clustering, dimension reduction, and focal/consensus CNA detection. Users can explore data through interactive heatmaps, phylogenetic trees, embedding plots, CN charts, and focal/consensus plots, or upload and annotate their own CNAs in real time. In all, with its large curated data volume and rich annotation capabilities, CNAScope serves as a vital resource for accelerating cancer research.
@article{CNAScope, title = {CNAScope: Pan-Cancer Copy Number Aberration Database with Functional Annotation and Interactive Visualization}, author = {Feng, Xikang and Zheng, Jieyi and Peng, Sisi and Jiang, Anna and Ng, Ka Ho and Lyu, Chengshang and Jin, Qiangguo and Chen, Lingxi}, journal = {Nucleic Acids Research}, pages = {D1364–D1375}, year = {2026}, publisher = {Oxford University Press}, doi = {10.1093/nar/gkaf1242}, peerreviewed = {true} } -
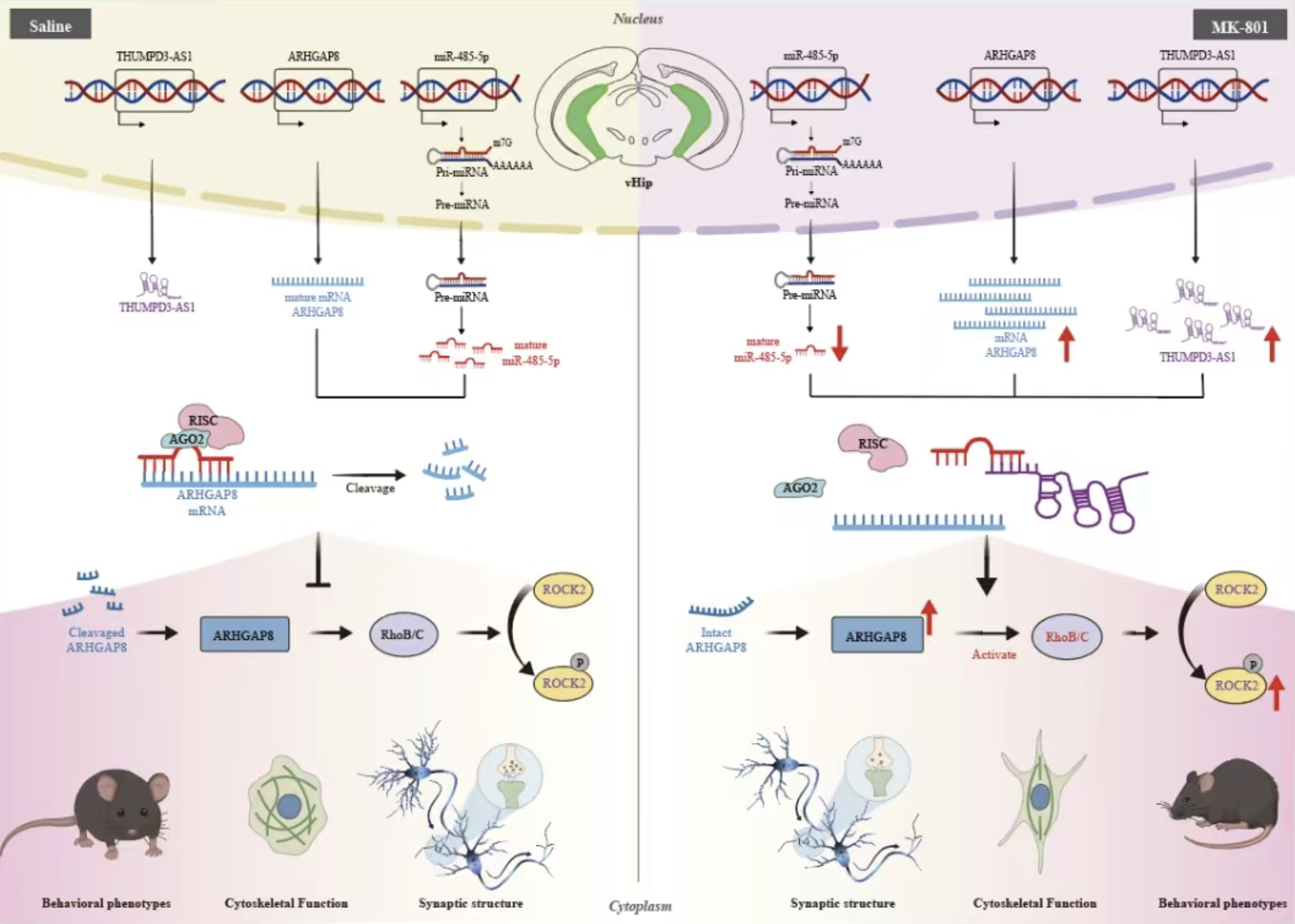 LncRNA THUMPD3-AS1 Regulates Behavioral and Synaptic Structural Abnormalities in Schizophrenia via miR-485-5p and ARHGAP8Xiaojuan Gong, Lingxi Chen, Xin Guo, Anna Jiang, Yayi He, Chunxia Yan, Liang Ma, Jiayang Gao, Jinyu Zhang, and Bao ZhangAdvanced Science, 2025
LncRNA THUMPD3-AS1 Regulates Behavioral and Synaptic Structural Abnormalities in Schizophrenia via miR-485-5p and ARHGAP8Xiaojuan Gong, Lingxi Chen, Xin Guo, Anna Jiang, Yayi He, Chunxia Yan, Liang Ma, Jiayang Gao, Jinyu Zhang, and Bao ZhangAdvanced Science, 2025Abstract Schizophrenia (SCZ) is characterized by synaptic structural deficits, yet how dysregulated noncoding RNAs (ncRNAs) drive these abnormalities remains unknown. Through integrative multilayered analysis of SCZ data from whole transcriptome sequencing (blood samples), GWAS risk loci, and expression data using pipeline ceRNAxis, the THUMPD3-AS1/miR-485-5p/ARHGAP8 axis is identified as a key regulator of synaptic function. Functional validation reveals that THUMPD3-AS1 acts as a competitive endogenous RNA, sequestering miR-485-5p and thereby derepressing ARHGAP8. Despite suppressing RhoA activity, ARHGAP8 enhances ROCK2 activation through RhoB/C-mediated compensatory mechanisms. Hyperactivation of ROCK2 through this noncanonical pathway disrupted actin cytoskeletal remodeling patterns, leading to increased immature dendritic spines and synaptic ultrastructural defects, which are pathological features associated with SCZ. In vivo, ventral hippocampal (vHip) overexpression of miR-485-5p or targeted knockdown of THUMPD3-AS1 rescued MK-801-induced SCZ-like phenotypes (anxiety, cognitive deficits, and social memory impairments) and restored synaptic ultrastructure. Crucially, this regulatory axis is cross-species conservation, with bidirectional expression changes validated in patient-derived blood and vHip tissues of mice. The findings reveal a novel ncRNA-driven pathogenic cascade in SCZ, where dysregulated RhoB/C-ROCK2 signaling, distinct from classical RhoA pathways, mediates synaptic destabilization. This presents a therapeutic axis for precision interventions targeting noncanonical actin cytoskeletal remodeling.
@article{ceRNAxis, author = {Gong, Xiaojuan and Chen, Lingxi and Guo, Xin and Jiang, Anna and He, Yayi and Yan, Chunxia and Ma, Liang and Gao, Jiayang and Zhang, Jinyu and Zhang, Bao}, title = {LncRNA THUMPD3-AS1 Regulates Behavioral and Synaptic Structural Abnormalities in Schizophrenia via miR-485-5p and ARHGAP8}, journal = {Advanced Science}, pages = {e08867}, year = {2025}, doi = {10.1002/advs.202508867}, peerreviewed = {true} }